IT-3-P-5986 Inherently coaligned dual color STED microscopy
Stimulated Emission Depletion (STED) Microscopy provides a versatile tool for investigating cellular structures on the nanoscale: While preserving the advantages of fluorescence microscopy, such as easy sample preparation and live cell imaging, the resolution is not limited by diffraction.
Here, we demonstrate the capabilities of STED microscopy for protein mapping and colocalization analysis. Applying a two color, pulse interleaved excitation and detection scheme combined with a single STED beam, two spectrally distinct dyes can be imaged almost simultaneously at a resolution down to 20nm. As the fluorescent volume in the sample is defined by the STED laser, the described setup provides a colocalization accuracy well below its resolution, insensitive to moderate misalignment and drift.
We imaged various proteins of the nuclear pore complex (Figure 1). Following standard immunolabeling protocols the staining was done with primary antibodies targeting the protein and secondary antibodies, tagged with the dye, targeting the primary antibody. Although the spectra of the dyes used for two-color imaging are overlapping, the crosstalk between the channels is below 20% and no post processing is required. These properties open the possibility to map even closely neighboring proteins, as exemplified on gp210 and NUP133.
Until recently, the lack of a suitable dye in the red wavelength region confined live cell STED imaging to wavelengths below 600nm. Long wavelengths however have the tendency to be less phototoxic and reduce background by autofluorescence. Here, we demonstrate live cell imaging with a newly developed silicon-rhodamine dye. Using the STED system described above we achieve a resolution better than 40nm in living cells, imaging microtubules and actin (Figure 2).
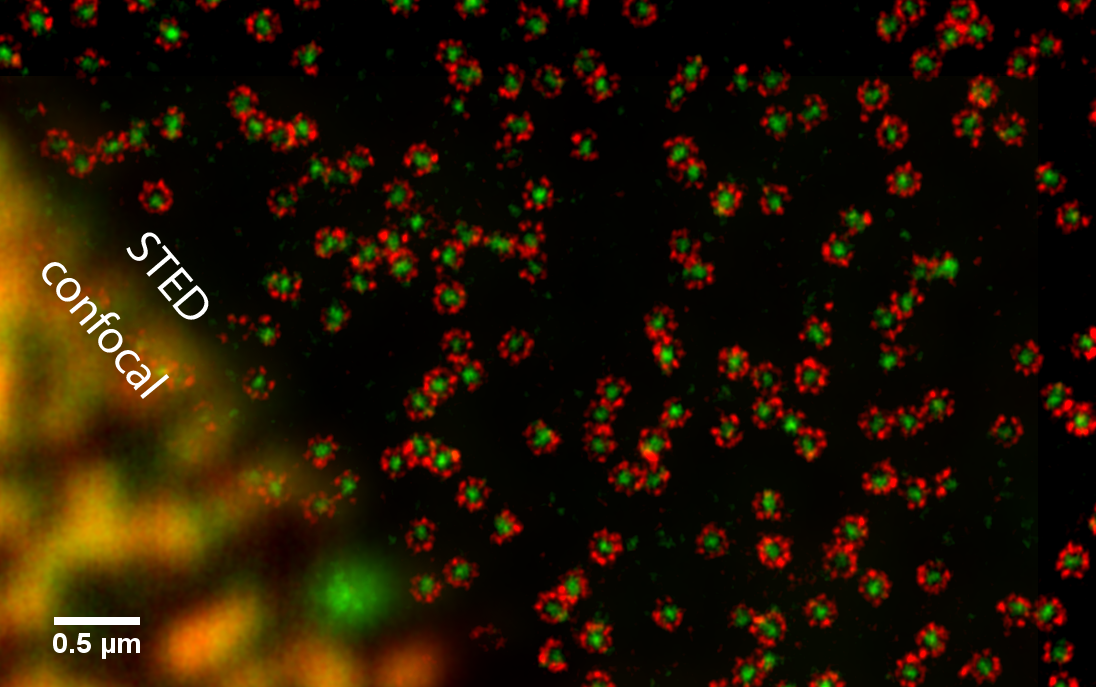
Fig. 1: STED image of nuclear pores in Xenopus cells. The protein gp210 (red) is arranged symmetrically around the central pore channel. As the diameter of the ring is approximately 160nm, the structure would not be resolvable with conventional fluorescence microscopy. In green, various FG repeat nucleoporins were labeled with a pan-specific antibody. |

Fig. 2: Live rat primary hippocampal neurons stained with the recently developed fluorogenic dye SiR-actin. Actin in axons can arrange in rings with a spacing of 180nm. The used silicon-rhodamine fluorophore is cell permeable and minimally toxic to the cell. Its high photostability makes it a versatile probe for superresolution microscopy. |