IT-9-P-1573 Charge Density Determination for Transition Metals and Intermetallics by Convergent Beam Electron Diffraction and Density Functional Theory Validation
The electron charge density difference, ∆ρ(r), i.e., the difference between the crystal electron density and that of the equivalent independent atom model (IAM), represents quantum mechanical characteristics central for fundamental understanding of materials. Convergent beam electron diffraction (CBED) permits probing of nano-scale volumes of perfect crystal and can enable measurements of low-order structure factors, Fg, with sufficient accuracy to obtain ∆ρ(r) for transition metals and binary intermetallic phases [e.g. 1-3]. The high accuracy and precision of the CBED measurements warrants their use as additional metrics in validation of density functional theory (DFT) calculations for these d-electron system materials [2]. Here, sets of multiple Fg and the Debye Waller factors have been determined simultaneously by CBED for transition metals (e.g. Cr, Fe, Ni, Co, Cu, Ta) and chemically ordered intermetallic phases (e.g. NiAl, TiAl, FePd). Using the local density approximation (LDA), LDA + U, and different generalized gradient approximations (GGA) functionals implemented in WIEN2K low-order Fg and thus ∆ρ(r) have been calculated for comparison with the CBED measurements. While many of the different GGA calculations achieve good overall agreement with the experimentally determined low-order Fg for the elements, LDA and GGA functionals fail to predict accurately the low-order Fg for β-NiAl and γ1-FePd. For equiatomic γ-TiAl GGA based DFT achieved considerably improved agreement with experimentally determined ∆ρ(r), when compared with LDA calculations [2]. Fig. 1 shows the difference between the X-ray structure factors, Fg, determined by CBED for two different composition TiAl crystals (Ti-50at%Al and Ti-52at%Al) and the IAM based Fg. Select data from ∆ρ(r)-maps obtained from CBED measurements and GGA DFT calculations are compared in Fig. 2 for the equiatomic and slightly Al-rich TiAl phases. Effects from the small (2at.%) Al-excess in the intermetallic γ-TiAl have been detected by the CBED experiments and are discernible in the ∆ρ(r) most clearly for the (001)-sections (Fig. 2). The excess Al is incorporated substitutionally on Ti sites and appears to enhance delocalization of charge density between second nearest neighbor Ti atoms along <010>, while reducing it for nearest neighbor Ti atom bonds along <110> (Fig. 2).
References
[1] XH Sang, AK Kulovits, JMK Wiezorek, Acta Crystal. A66 (2010) p. 694
[2] XH Sang et al., J. Chem. Phys. 138 (2013) p.084504
[3] XH Sang, et al., Phil. Mag. 92 (2012) p.4408
The authors acknowledge support from the Office of Basic Energy Sciences, Division of Materials Science and Engineering (Grant No. DE-FG02-08ER46545).
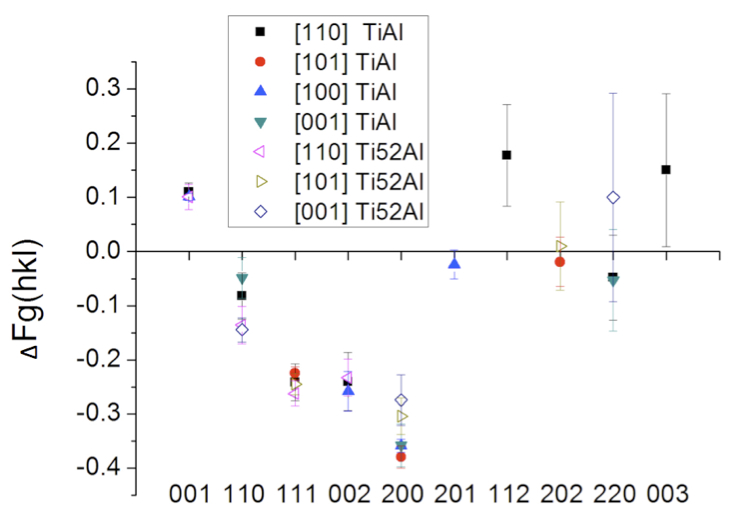
Fig. 1: Fig. 1: Difference between the CBED determined X-ray structure factors and the IAM structure factors, ∆Fg, for equiatomic (TiAl) and Al-rich off-stoichiometric (Ti-52Al) γ-TiAl phase. The hkl are plotted along the abscissa. The [uvw] in the legend (inset) indicate the approximate incident beam direction in CBED experiments. |
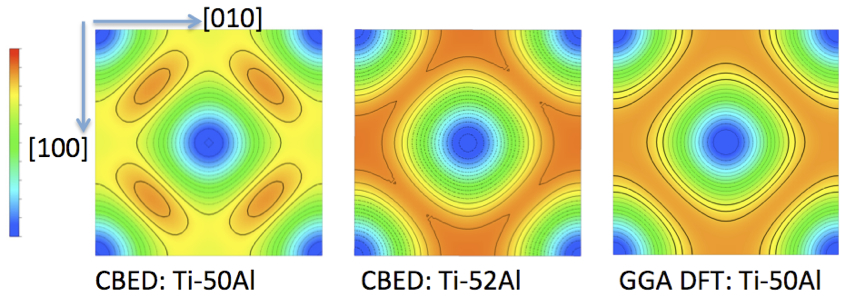
Fig. 2: Fig. 2: Example ∆ρ(r) sections in (001), all Ti plane for the equiatomic composition phase, of L1o-structure tP4 unit cell of TiAl. CBED derived for Ti-50Al (equiatomic) on the left, CBED derived for Ti-52Al (Al-rich) in the middle, and DFT calculated for Ti-50Al (equiatomic) on the right. |